
Полный научный перевод статьи Hervé Puy, Laurent Gouya, Jean-Charles Deybach
Наследственные порфирии: нарушения метаболизма гема.
Восемь наследственных порфирий являются заболеваниями, каждое из которых связано с поражением одного из ферментов биосинтеза гема. Они характеризуются острыми нейровисцеральными симптомами и/или поражениями кожи. Эти клинические признаки являются следствием накопления предшественников гема. Семь порфирий являются следствием частичного дефицита ферментативной активности; напротив, одна недавно описанная порфирия является следствием приобретения функции (gain-of-function). Острые порфирии проявляются интермиттирующими нейровисцеральными кризами, типично сочетающими сильные боли в животе, тошноту, запор, спутанность сознания, а иногда осложняются судорожными приступами и тяжелыми неврологическими нарушениями, которые могут угрожать жизни. Кожные порфирии проявляются либо в виде болезненной фотосенсибилизации, либо в виде фотодерматоза с хрупкостью кожи, сопровождающейся буллезными высыпаниями. В последние годы структурирование на европейском уровне сети центров экспертизы позволило достичь значительного прогресса в понимании патофизиологии и оригинального типа генетической передачи этих заболеваний, что способствовало значительному улучшению диагностики и ведения пациентов и их семей.
КРАТКОЕ РЕЗЮМЕ
Наследственные порфирии представляют собой группу из восьми метаболических нарушений пути биосинтеза гема, характеризующихся острыми нейровисцеральными симптомами и/или поражениями кожи. Каждая порфирия вызвана дисфункцией определенного ферментативного этапа, что приводит к специфическому накоплению предшественников гема. Семь порфирий обусловлены частичным дефицитом фермента, в то время как механизм «приобретения функции» недавно был идентифицирован при новой порфирии. Острые порфирии проявляются сильной абдоминальной болью, тошнотой, запором и спутанностью сознания, а иногда осложняются судорогами и тяжелыми неврологическими расстройствами, которые могут быть жизнеугрожающими. Кожные порфирии также могут проявляться либо острой болезненной фотосенсибилизацией, либо хрупкостью кожи и волдырями. Редкие рецессивные порфирии обычно манифестируют в раннем детстве либо тяжелыми хроническими неврологическими симптомами, либо хроническим гемолизом и тяжелой кожной фотосенсибилизацией. Порфирии по-прежнему недостаточно диагностируются, но последние достижения в изучении патогенеза и генетики порфирий у человека ведут к улучшению помощи этим пациентам и их семьям.
ВВЕДЕНИЕ
Порфирии — это группа из 8 наследственных метаболических заболеваний, поражающих биосинтез гема [1]. Каждая из них является результатом специфического ферментативного дефекта в метаболической цепи (рис. 1). Специфические профили накопления предшественников гема: 8-аминолевулиновой кислоты (АЛК), порфобилиногена (ПБГ) и/или порфиринов — ассоциированы с характерными клиническими проявлениями: острыми нейровисцеральными атаками, поражениями кожи или сочетанием обоих типов [1].
МЕТАБОЛИЗМ ГЕМА И КЛАССИФИКАЦИЯ ПОРФИРИЙ
Начиная с глицина и сукцинил-КоА в митохондриях, синтезирующих АЛК, цепь продолжается тремя цитоплазматическими этапами до заключительных реакций образования гема, которые вновь протекают в митохондриях (рис. 1). Гем необходим для жизни всех клеток человека, участвуя, в частности, в реакциях дыхания и окислительно-восстановительных реакциях, однако большая его часть продуцируется в эритропоэтических клетках (для синтеза гемоглобина) и в паренхиматозных клетках печени (для синтеза цитохромов и гемопротеинов). Регуляция продукции гема различается между этими двумя тканями, главным образом, из-за разницы в скорости начального синтеза АЛК [1]. Первый фермент, 8-аминолевулинатсинтаза (АЛАС, EC 2.3.1.37), кодируется двумя генами: один — эритроспецифичный (ALAS2 на Х-хромосоме, кодирует фермент АЛАС2), другой — с убиквитарной экспрессией (ALAS1 на хромосоме 3, кодирует АЛАС1). АЛАС1 катализирует лимитирующую стадию синтеза гема в печени; уровень его активности зависит от регуляции по механизму отрицательной обратной связи, осуществляемой пулом внутриклеточного так называемого свободного или несвязанного гема (рис. 2).
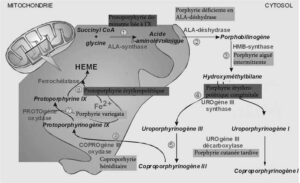
Рис. 2. — Печеночная регуляция биосинтеза гема: центральная роль конечного продукта — гема.
*СО: монооксид углерода; Cyt: цитохром; ALAS: 8-аминолевулинатсинтаза.*
В клетках эритроцитарного ряда синтез гема регулируется в ходе эритроидной дифференцировки в ответ на эритропоэтин. В этих клетках активность АЛАС2 не подвергается отрицательной обратной регуляции со стороны гема, но продукция гема ограничена доступностью железа [1-3]. Макрофаги селезенки и печени деградируют гем и рециркулируют железо, полученное при фагоцитозе эритроцитарных клеток, с помощью индуцибельной гемоксигеназы 1 (HO-1, рис. 2) [4].
Порфирии часто классифицируют в зависимости от их клинических проявлений на острые порфирии (ОП), кожные порфирии и несколько редких рецессивных порфирий (рис. 1; Таблица 1), а также в зависимости от ткани, в которой преобладает избыточная продукция порфиринов и их предшественников АЛК и ПБГ, в основном печени и костного мозга. Таким образом, различают печеночные порфирии и эритропоэтические порфирии.
ОСТРЫЕ ПОРФИРИИ
Острые порфирии (ОП) с аутосомно-доминантным типом наследования: острая интермиттирующая порфирия (ОИП, MIM 176000), пестрая порфирия (ПП, MIM 176200) и наследственная копропорфирия (НКП, MIM 121300) — являются печеночными порфириями, которые могут угрожать жизни во время острых интермиттирующих кризов с нейровисцеральной симптоматикой [1]. Эти острые атаки редки и трудны для диагностики из-за низкой пенетрантности и вариабельной клинической экспрессивности как у разных пациентов, так и у одного и того же пациента. Кожные поражения никогда не наблюдаются при ОИП, но могут быть единственным клиническим проявлением ПП (60% случаев) и, реже, НКП. ОИП поражает примерно одного человека на 100 000 во Франции (см. раздел D ниже). Распространенность ПП составляет примерно половину от таковой при ОИП. Острые кризы чаще встречаются у женщин (80% случаев), исключительны до пубертата и реже после менопаузы, достигая пика частоты у лиц в возрасте около 30 лет. Большинство пациентов переносят один или несколько кризов с последующей полной ремиссией на всю оставшуюся жизнь; менее 10% будут иметь рецидивирующие острые кризы.
A. Клиническая картина
Кризы ОП начинаются с продромальной фазы, включающей незначительные изменения поведения, такие как тревога, бессонница и невозможность найти покой. Большинство этих атак начинается с сильных болей в животе. Тошнота, рвота и запор часты, как и тахикардия, гипергидроз или артериальная гипертензия — все симптомы симпатической нейровегетативной гиперактивности. Клиническое обследование не выявляет отклонений, и радиологические данные обычно нормальны. Во время этих острых фаз часто возникают обезвоживание и гидроэлектролитные нарушения. В 40% случаев может наблюдаться гипонатриемия вследствие неадекватной секреции АДГ, приводящая к судорогам в тяжелых случаях. Красный или темный цвет мочи, хотя и непостоянный, может помочь врачам в их исследованиях. У 20-30% пациентов возможно наблюдать психические нарушения, такие как тревога, депрессия, дезориентация во времени и пространстве, галлюцинации, паранойя или спутанные состояния. В меньшей степени может наблюдаться мышечная слабость вплоть до паралича.
Эти острые кризы обычно длятся от 1 до 2 недель. Если они затягиваются, то обычно приводят к значительной потере веса, но именно тяжелые неврологические осложнения угрожают жизни. В частности, нейропатия часто возникает, когда во время криза используются лекарственные средства, известные как порфириногенные. Это обычно моторная нейропатия, начальные проявления которой очень часто включают боли в конечностях («мышечные боли») и слабость, начинающуюся в проксимальных мышцах. Эта мышечная слабость может усилиться до тетраплегии с бульбарным и дыхательным параличом, что может привести к смерти. Если такой паралич восстанавливается, то постепенно, иногда неполностью, с последствиями, локализующимися в основном в конечностях. Также могут существовать пирамидные знаки, мозжечковый синдром, транзиторная слепота или нарушения сознания. Цереброспинальная жидкость (ЦСЖ) чаще всего нормальна. Важно отметить, что в настоящее время, благодаря улучшению специализированной помощи (Центр экспертизы редких заболеваний CRMR Porphyries), семейному скринингу пресимптоматических носителей и лучшей осведомленности специалистов, порфириновая нейропатия встречается гораздо реже, чем в прошлом, а острые атаки исключительно редко заканчиваются летально.
B. Параклинические исследования. Диагностика острого криза
Поскольку клинические проявления в большинстве случаев неспецифичны, биохимические анализы необходимы для положительной диагностики острого криза, а также для определения типа порфирии.
Исследование избытка ПБГ в моче является основным анализом первой линии у пациентов с подозрением на атаку острой порфирии. Измерение АЛК не является обязательным для установления диагноза, но может помочь дифференцировать его от других метаболических причин болей в животе, таких как отравление свинцом или крайне редкая АЛК-дегидратазная порфирия (АДП). Уровни ПБГ и АЛК в моче повышены при всех трех ОП (ОИП, ПП и НКП), хотя повышение более выражено и длится дольше при ОИП [3]. Измерение порфиринов в моче не представляет интереса. При значительном и документированном повышении экскреции ПБГ с мочой лечение должно быть начато немедленно, в то время как высокоспециализированные анализы позволят определить тип порфирии (Таблица 1).
Таблица 1. Биохимическая диагностика и скрининг носителей при порфириях
Примечание: Полный перевод таблицы требует отдельного форматирования, но ключевые данные:
-
*ОИП: Моча — ПБГ, АЛК, Уро III; Активность HMBS 50%; Секвенирование гена HMBS.*
-
*НКП: Моча — ПБГ, АЛК, Копро III; Кал — Копро III (отношение изомеров III/I >2,0); Секвенирование CPOX.*
-
*ПП: Моча — ПБГ, АЛК, Копро III; Кал — Прото IX > Копро III; Плазма — пик 624-627 нм; Секвенирование PPOX.*
-
*ЭПП: Эритроциты — свободный Прото IX; Плазма — пик 630-634 нм; Секвенирование FECH + аллель IVS3-48C.*
-
*Х-сцепленная доминантная ПП: Эритроциты — Zn-Прото IX и свободный Прото IX; Секвенирование ALAS2.*
-
СПК: Моча — Уро I/III, Гепта; Кал — Изокопро, Гепта; (для семейной формы — снижение активности UROD).
*ALA: 8-аминолевулиновая кислота; PBG: порфобилиноген; Uro: уропорфирин; Copro: копропорфирин; Proto: протопорфирин; Isocopro: изокопропорфирин; Hepta: гептакарбоксипорфирин; ND: не определено; I или III: тип изомеров.*
Эти исследования должны проводиться в связи с Центром экспертизы редких заболеваний «Порфирии» (CRMR Porphyries).
Диагностика типа острой порфирии у пациента в кризе
Спектрометрия флуоресценции плазмы пациентов является анализом первой линии, поскольку пик на 624-628 нм устанавливает диагноз ПП. Однако он не дифференцирует ОИП и НКП. Анализ только порфиринов мочи недостаточно дискриминативен (Таблица 1). Концентрация общих порфиринов кала повышена при ПП с более высокой концентрацией протопорфирина (ППIX), чем копропорфирина, в то время как при ОИП она обычно нормальна [1]. При НКП концентрация общих порфиринов кала в большинстве случаев повышена, это повышение касается в основном копропорфирина с отношением изомеров III/I > 2,0 (Таблица 1). Когда она присутствует, 50% снижение активности ПБГ-деаминазы (ПБГД или гидроксиметилбилансинтазы, HMBS) позволяет идентифицировать пациентов с ОИП.
Диагностика в период ремиссий и семейные исследования
В период ремиссий концентрации порфиринов в моче, кале или плазме могут быть нормальными при всех трех ОП. Наиболее чувствительным тестом для ПП в ремиссии или до появления симптомов является спектрометрия флуоресценции плазмы, если пациент старше 15 лет. Для диагностики НКП отношение изомеров III/I копропорфиринов кала > 2,0 является чувствительным у взрослых [31].
Скрининг в семьях необходим для предотвращения возникновения острых кризов у пресимптоматических носителей. Анализ ДНК для поиска мутации гена, ответственного за заболевание в семье, стал золотым стандартом [5-9]. Он требует обязательного предварительного выявления мутации у пораженного члена семьи. Гены всех порфирий охарактеризованы, и идентифицировано большое количество мутаций, специфичных для каждой патологии. Регулярно обновляемые списки мутаций доступны на сайте базы данных «Human Gene Mutation Database» (www.hgmd.cf.ac.uk). Ферментативные измерения зарезервированы для семей, в которых мутация еще не идентифицирована (Таблица 1). В любом случае, измерение активности протопорфириногеноксидазы и копропорфириногеноксидазы, а также более распространенной ПБГД, должно проводиться в референсном центре по порфириям (CRMR Porphyries, www.porphyrie.net).
C. Порфириновая нейропатия и патофизиология острых порфириновых кризов
Порфириновая нейропатия
Все клинические проявления острой атаки могут быть объяснены поражением нервной системы. Порфириновая нейропатия представляет собой первичное поражение нейрона, метаболическую аксонопатию, которая постепенно приводит к гибели нейронов. Это нейрональное поражение хорошо коррелирует с наблюдаемыми симптомами и клиническими признаками и затрагивает одновременно, но в разной степени у каждого пациента, три нервные системы: периферическую, вегетативную и центральную [38].
-
Периферическая нервная система: Аксональная дегенерация является первым событием с поражением волокон на всех стадиях дегенерации и одновременно признаками тонкой регенерации. Структурный анализ на ранних стадиях порфириновой нейропатии выявляет паранодальную ретракцию миелина, линейно распределенные миелиновые шары и выраженную аксональную дегенерацию. Электронная микроскопия волокон позволяет заключить, что первичной является аксональная дегенерация с последующей демиелинизацией (рис. 4).
-
Вегетативная нервная система: В немногих сообщениях отмечались хроматолиз нейронов спинальных ганглиев, нейронов серого вещества спинного мозга и ядер ствола мозга, дегенерация волокон спинного мозга и мозжечка, но редко демиелинизация. Преимущественно поражаются парасимпатические волокна с аксональной дегенерацией и демиелинизацией [40].
-
Центральная нервная система: Немногие МРТ головного мозга, выполненные во время острого криза, показывают в основном диффузные мультифокальные ишемические поражения, ассоциированные с микроинфарктами — аномалии, которые могут быть обусловлены вазоспазмом, индуцированным кризом.
Патофизиология острых кризов
Предполагается, что гиперпродукция АЛК печенью нейротоксична. Но снижение синтеза гема, особенно в нервной ткани, также может объяснять некоторые проявления порфириновой нейропатии.
-
Доказательства в пользу основной роли АЛК в нейротоксичности:
-
Острый порфириновый криз биологически характеризуется повышением активности АЛАС1 и сильно увеличенной продукцией/экскрецией АЛК.
-
Лечение, считающееся эффективным при ОИП (гем и высокие дозы глюкозы), снижает активность АЛАС1 и накопление АЛК.
-
При других заболеваниях (тирозинемия типа I, отравление свинцом, дефицит АЛК-дегидратазы) наблюдается высокая экскреция АЛК и сходные клинические признаки (абдоминальная боль, периферическая нейропатия) [1].
-
АЛК структурно близка к ГАМК и может нарушать функцию ГАМК-ергической системы. АЛК также является структурным аналогом глутамата [45].
-
Трансплантация печени у пациентов с порфирией полностью устраняет кризы и нормализует АЛК/ПБГ, тогда как домино-трансплантация индуцирует симптомы [10].
-
-
Доказательства против роли АЛК в нейротоксичности:
-
Многие бессимптомные носители экскретируют высокие уровни АЛК/ПБГ, сходные с симптомными пациентами.
-
Внутривенное введение АЛК здоровым добровольцам не вызвало неврологических аномалий [43].
-
У мышей с дефицитом ПБГ-деаминазы периферическая нейропатия развивается без увеличения продукции/экскреции АЛК [42].
-
-
Доказательства потенциальной патогенной роли дефицита гема:
-
Гем модулирует экспрессию генов в нейронах.
-
Его дефицит снижает активность триптофанпирролазы, увеличивая триптофан и серотонин, что может вызывать изменения настроения и кишечный паралич [46].
-
Дефицит гема может нарушать путь гемоксигеназа — СО — цГМФ в его антиноцицептивной роли, снижая болевой порог [41].
-
D. Лечение острого порфиринового криза
Острые атаки вызываются факторами, которые либо напрямую индуцируют АЛАС1, либо увеличивают потребность в синтезе гема в печени [11], и, следовательно, дерепрессируют АЛАС1 (рис. 2). Эти факторы включают гормональные колебания менструального цикла, голодание, курение, инфекции и воздействие порфириногенных лекарств [12-13].
Поддерживающая терапия:
Часто требуются высокие дозы опиатов в сочетании с антиэметиком и фенотиазином. Необходимо тщательно следить за гидроэлектролитным балансом, особенно из-за риска тяжелой гипонатриемии. Важно обеспечить достаточное поступление калорий, перорально богатыми углеводами. При сердечно-сосудистых осложнениях (гипертензия, тахикардия) могут потребоваться бета-блокаторы [15]. При развитии моторной нейропатии и снижении жизненной емкости легких из-за паралича межреберных мышц необходима искусственная вентиляция легких.
Специфическое лечение:
Внутривенное введение гемина (гема аргинат) [15], который подавляет АЛАС1 и снижает экскрецию АЛК и ПБГ с мочой, является лечением выбора. Большинство пациентов с неосложненными атаками улучшаются в течение первых 5 дней. Однако гемин не вызывает регресса уже установленной нейропатии, но может предотвратить ее появление и замедлить дальнейшее ухудшение, если введен достаточно рано. Стабильный препарат с раствором гемина, комплексированного с аргинином (Нормосанг®, Orphan Europe), широко доступен в Европе. Побочные эффекты при краткосрочном лечении редки.
Рецидивирующие острые атаки (менее 10% пациентов):
Лечение сложное, требует длительной терапии гемином. Регулярное введение (иногда раз в неделю) может помочь контролировать болезнь [1]. У этих пациентов может потребоваться постановка постоянного венозного катетера. Следует учитывать риск перегрузки железом (одна доза гемина содержит 22,7 мг железа). Трансплантация печени должна рассматриваться у пациентов с наиболее тяжелыми формами ОИП [10].
Профилактика и наблюдение при острых порфириях:
Носители генетического дефекта должны получать активную профилактику и строгие гигиено-диетические рекомендации: нормальное питание с регулярными приемами пищи, избегать алкоголя и табака, консультироваться со списком безопасных/небезопасных лекарств (www.porphyria-europe.org) [14]. Ранняя и точная диагностика, а также эффективные консультации и лечение значительно снизили смертность от ОП [3; 17]. У пациентов (как симптомных, так и с латентной болезнью) повышен риск развития артериальной гипертензии, гепатоцеллюлярной карциномы и хронической почечной недостаточности [18-19].
E. Острая интермиттирующая порфирия во Франции в 2014 году: генетические и эпидемиологические аспекты
ОИП — аутосомно-доминантное заболевание, вызванное мутациями в гене HMBS (11q23.3). По состоянию на 15 сентября 2015 года в мире идентифицировано 396 различных мутаций. За 40 лет деятельности Французский центр порфирий (CRMR Porphyries) зарегистрировал почти 600 пациентов с ОИП из 370 семей. Распространенность заболевания во Франции очень низкая, около 1/100 000, заболеваемость составляет около 5-15 новых случаев в год. Распространенность мутантного гена в общей популяции гораздо выше — около 1/1000 (данные Exome Variant Server), что позволяет оценить пенетрантность заболевания в общей популяции Франции менее 1% (0,0025). Напротив, анализ 370 родословных семей с ОИП позволил оценить среднюю пенетрантность внутри семей примерно в 30%, что предполагает наличие дополнительных генетических, эпигенетических или средовых факторов, влияющих на фенотипическую экспрессию.
КОЖНЫЕ ПОРФИРИИ
Буллезные порфирии
Клинические симптомы:
Пестрая порфирия (ПП), наследственная копропорфирия (НКП) и кожная порфирия (КП, поздняя кожная порфирия) имеют сходные кожные проявления. КП (печеночная, но не острая порфирия) является наиболее частой порфирией в мире и проявляется исключительно кожными знаками [20]. Поражения кожи ограничены участками, подверженными воздействию солнца (тыльная поверхность кистей, лицо, шея, у женщин — ноги и верхняя часть стоп). Хрупкость кожи — самый специфичный элемент: на минимальную травму возникает поверхностная эрозия, быстро покрывающаяся коркой. Пузыри или везикулы заживают несколько недель, оставляя рубцы и милиумы. Часто наблюдается гипертрихоз и гиперпигментация. Различная степень поражения печени часто встречается у пациентов с КП, особенно в сочетании с чрезмерным употреблением алкоголя [1].
Патогенез поражения кожи:
В коже накапливаются большие количества порфиринов печеночного происхождения. Тетрапиррольное ядро порфиринов делает их высокофотосенсибилизирующими; они поглощают световую энергию в видимом спектре около 400 нм [1]. Возбужденные молекулы порфиринов передают энергию биологическим молекулам, вызывая перекисное окисление липидов мембран или окисление нуклеиновых кислот и полипептидов.
Биохимическая диагностика:
Флуоресцентный спектр плазмы является лучшим первоначальным тестом для диагностики кожных порфирий, дифференцируя ПП и КП. У пациентов с симптоматической КП, помимо высокой экскреции уропорфирина и 7-карбоксипорфирина, основным порфирином, экскретируемым с калом, является изокопропорфирин (Таблица 1).
КП обусловлена дефицитом активности уропорфириногендекарбоксилазы (УРОД) по крайней мере в печени (рис. 1). Спорадический подтип (sPCT, 75% случаев, MIM 176090) чаще наблюдается у мужчин без семейного анамнеза; активность УРОД снижена только в печени [20]. Семейный подтип (fPCT, 25% случаев, MIM 176100) имеет более раннее начало, аутосомно-доминантное наследование с низкой пенетрантностью, обусловлен мутациями гена UROD и конститутивным 50% дефицитом активности УРОД. Активность УРОД в эритроцитах нормальна при sPCT и снижена при fPCT.
Факторы риска:
Злоупотребление алкоголем, прием эстрогенов, гепатит С, в меньшей степени ВИЧ-инфекция и генетический гемохроматоз. Они действуют сами по себе или в сочетании с перегрузкой печени железом, которая генерирует железо-зависимый окислительный механизм. Аллели C282Y и H63D гена HFE ассоциированы с повышенным риском КП [1].
Лечение и наблюдение:
Исключить алкоголь, избегать солнца. При КП без гемохроматоза широко используется хлорохин в низких дозах (100 мг 2 раза в неделю). Кровопускания являются лечением выбора для пациентов с КП и гемохроматозом (одна единица крови 350-500 мл еженедельно до нормализации запасов железа). Клиническая ремиссия достигается через 6-9 месяцев. У пациентов с КП и ХПН лечение эритропоэтином позволяет уменьшить избыточные запасы железа.
Острые болезненные фотосенсибилизирующие порфирии
Эритропоэтическая протопорфирия (ЭПП)
-
Клиническая картина и диагностика:
ЭПП (MIM 177000) — наследственное нарушение, вызванное частичным дефицитом митохондриальной феррохелатазы (FECH) [21]. Накопление свободного ППIX в эритроцитах, а затем и в других тканях, сопровождается инвалидизирующей болезненной фотосенсибилизацией и, реже, потенциально тяжелыми осложнениями со стороны печени [22]. Симптомы: жжение, покалывание и зуд на открытых участках кожи вскоре после воздействия солнца. У <2% пациентов может развиться быстро прогрессирующее холестатическое поражение печени с печеночно-клеточной недостаточностью.
Тип наследования сложный: у 94% пациентов требуется котрансмиссия частной мутации FECH в транс-положении с гипоморфным аллелем *FECH IVS3-48C*, который снижает остаточную активность FECH ниже критического порога (~35%) [24-25].
Диагностика: массивное повышение свободного ППIX в эритроцитах. Флуоресценция плазмы: характерный пик при 634 нм. Активность FECH снижена до 10-35% от нормы. -
Лечение:
Защита от солнечного света. Альфа-меланотид (аналог α-МСГ) стимулирует фоточувствительную выработку меланина. Пероральный бета-каротин эффективен менее чем у 1/3 пациентов. При тяжелом поражении печени — трансплантация печени. Во время операции необходима защита от фототоксического повреждения внутренних органов (физический барьер, желтый светофильтр) [21]. Возможна сочетанная трансплантация печени и костного мозга для предотвращения рецидива поражения печени [1].
Х-сцепленная доминантная протопорфирия (XLDPP)
Недавно описанная форма [28], клинически очень сходная с ЭПП, с высоким уровнем внутриэритроцитарного ППIX (около 40% связано с цинком), но без дефицита феррохелатазы. Вызвана приобретением функции (gain-of-function) ALAS2 (делеции C-конца) [29], что приводит к избыточной продукции ППIX по сравнению с гемоглобином [30]. Лечение аналогично ЭПП.
РЕДКИЕ РЕЦЕССИВНЫЕ ПОРФИРИИ
A. Врожденная эритропоэтическая порфирия (ВЭП, болезнь Гюнтера)
Аутосомно-рецессивное наследование, обусловлена выраженным дефицитом активности уропорфириноген-III-синтазы (UROS) [31]. Клинически: тяжелая фотосенсибилизация, приводящая к пузырям, рубцам, мутиляции (потеря пальцев), и хронический гемолиз. Эритродонтия (красное окрашивание зубов). Красная флуоресценция мочи в пеленках. Аллогенная трансплантация костного мозга — единственное радикальное лечение [32]. Основа поддерживающей терапии — экстремальная фотозащита.
B. Гепатоэритропоэтическая порфирия (ГЭП)
Крайне редкая порфирия, обусловленная гомозиготным или компаунд-гетерозиготным дефицитом УРОД. Клинически напоминает ВЭП (детство, красная моча, пузыри, гипертрихоз, рубцы), иногда с гемолитической анемией и спленомегалией. Кровопускания и хлорохин неэффективны.
C. Редкие гомозиготные острые печеночные порфирии (АДП, ОИП, ПП, НКП)
Проявляются в неонатальном периоде или раннем детстве аномальным оранжевым/красноватым окрашиванием пеленок. Клинические фенотипы тяжелые: задержка психомоторного развития, неврологические расстройства без болей в животе (ОИП); поражения кожи + аномалии скелета (ПП). При гомозиготной НКП описаны два типа: 1) с задержкой развития и фотосенсибилизацией; 2) хардеропорфирия — с рождения интенсивная желтуха и гемолитическая анемия без неврологических симптомов [37]. АЛК-дегидратазная порфирия (АДП): крайне редка (6 описанных случаев), тяжелая неврологическая симптоматика (хроническая нейропатия, острые кризы), массивная экскреция АЛК и копропорфирина с мочой, активность АЛАД в эритроцитах резко снижена. Лечение как при острых кризах.
